

Data presented at TCT Connect, the 32nd annual scientific symposium of the Cardiovascular Research Foundation, finds early identification of right heart failure and early use of Impella RP is associated with significantly higher survival rates. Early identification of patients requiring right-heart support is critical because prior studies have shown 37% of AMI cardiogenic shock (AMICS) patients exhibit right heart dysfunction1, which results in an eight times increased risk of mortality2.
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20201016005123/en/
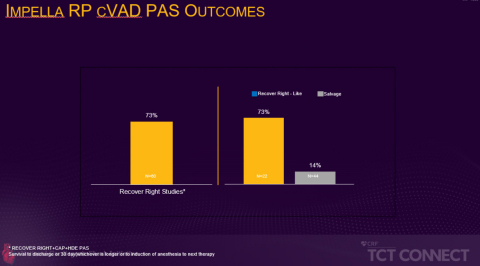
Figure 1 (Graphic: Business Wire)
The prospective, multi-center FDA PMA post-approval study presented at TCT compared survival in patients who would have met enrollment criteria for the RECOVER RIGHT trial to those who would not have qualified for the trial because they were in cardiogenic shock for more than 48 hours. The RECOVER RIGHT trial and subsequent HDE post-approval study data were collected between 2012 and 2017 and led to a PMA approval for the Impella RP in 2017. The ongoing PMA post-approval study presented at TCT Connect enrolled patients from September 2017 through June 2019 and found patients who received Impella RP support within 48 hours of cardiogenic shock onset had a significantly higher survival rate than those who received delayed right-heart support (73% vs. 14%, p<0.001). The 73% survival rate is comparable to the pre-PMA RECOVER RIGHT and HDE studies survival rate. (see figure 1)
“Early detection of right heart failure and early action is key to improving patient survival rates,” said Mark Anderson, MD, chair of the Department of Cardiac Surgery at HUMC/Hackensack Meridian Health. “This study suggests that in right heart failure, reducing the time between shock onset and initiation of Impella RP is a key element of proper patient support.”
A second study presented at TCT aims to help clinicians identify early triggers of right heart failure. The analysis of 100 patients performed by lead investigators of the National Cardiogenic Shock Initiative (NCSI) Study compared AMICS patients with right ventricular failure (RVF) to those without and found persistent diastolic suction alarms on the Automated Impella Controller (AIC) and an elevated central venous pressure (CVP) of greater than 12mmHg can be an early indication of RVF. (see figure 2)
“The real-time insight from the AIC can be an important tool to help a physician achieve better patient outcomes,” said Babar Basir, DO, interventional cardiologist at Henry Ford Hospital. “Prolonged diastolic suction alarms can be an early marker of right ventricular failure in patients with elevated filling pressures, and increased duration of diastolic suction is associated with worse outcomes.”
Impella RP is the most studied right-sided device and the only percutaneous technology with FDA approval designating it as safe and effective for right-heart support. Its exclusive FDA approval is a result of five years of research that included:
- RECOVER RIGHT, an FDA-approved, prospective, multicenter, single-arm study, which commenced after the company received FDA investigational device exemption (IDE) approval in November 2012 and concluded in 2014
- A Continuous Access Protocol (CAP)
- HDE post-approval study, which was completed in 2017
- PMA post-approval study, initiated in September 2017
In addition, on May 29, 2020, the FDA issued an Emergency Use Authorization (EUA) to expand the use of Impella RP to include patients suffering from COVID-19-related right ventricular complications, including right ventricular dysfunction associated with pulmonary embolism. Impella is the only cardiovascular therapeutic device that has received FDA emergency use authorization to treat COVID-19 patients.
________________
1 Lala et al. J Card Fail. 2018;24:148-156
2 Mehta et al. J Am Coll Cardiol. 2001;37:37-43
ABOUT IMPELLA HEART PUMPS
The Impella 2.5® and Impella CP® devices are U.S. FDA PMA approved to treat certain advanced heart failure patients undergoing elective and urgent percutaneous coronary interventions (PCI) such as stenting or balloon angioplasty, to reopen blocked coronary arteries. The Impella 2.5, Impella CP, Impella CP with SmartAssist®, Impella 5.0®, Impella LD®, and Impella 5.5® with SmartAssist® are U.S. FDA approved heart pumps used to treat heart attack or cardiomyopathy patients in cardiogenic shock, and have the unique ability to enable native heart recovery, allowing patients to return home with their own heart. The Impella RP® is U.S. FDA approved to treat right heart failure or decompensation following left ventricular assist device implantation, myocardial infarction, heart transplant or open-heart surgery. The Impella RP is also authorized for emergency use by healthcare providers (HCPs) in the hospital setting for providing temporary right ventricular support for up to 14 days in critical care patients with a body surface area ≥1.5 m2, for the treatment of acute right heart failure or decompensation caused by complications related to coronavirus disease 2019 (COVID-19), including pulmonary embolism (PE). The Impella RP has not been cleared or approved for the treatment of acute right heart failure or decompensation caused by complications related to COVID-19. Impella Left Ventricular (LV) Support Systems are also authorized for emergency use by HCPs in the hospital setting for providing temporary (≤ 4 days for Impella 2.5, Impella CP, and Impella CP with SmartAssist; and ≤ 14 days for Impella 5.0 and Impella 5.5 with SmartAssist) LV unloading and support to treat critical care patients with confirmed COVID-19 infection who are undergoing ECMO treatment and who develop pulmonary edema while on V-A ECMO support or late cardiac decompensation from myocarditis while on V-V ECMO support. The authorized Impella LV Support Systems have neither been cleared or approved for the authorized indication for use. The Impella RP and Impella LV Support Systems have been authorized for the above emergency use by FDA under an EUA and have been authorized only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of medical devices under section 564(b)(1) of the Act, 21 U.S.C. § 360bbb-3(b)(1), unless the authorization is terminated or revoked sooner.
In Europe, the Impella 2.5, Impella CP and Impella CP with SmartAssist are CE marked for treatment of high-risk PCI and AMI cardiogenic shock patients for up to 5 days. Impella 5.0 and Impella LD are CE marked to treat heart attack or cardiomyopathy patients in cardiogenic shock for up to 10 days. The Impella 5.5 with SmartAssist is CE marked to treat heart attack or cardiomyopathy patients in cardiogenic shock for up to 30 days. The Impella RP is CE marked to treat right heart failure or decompensation following left ventricular assist device implantation, myocardial infarction, heart transplant, open-heart surgery, or refractory ventricular arrhythmia. To learn more about the Impella platform of heart pumps, including their approved indications and important safety and risk information associated with the use of the devices, please visit www.impella.com.
ABOUT ABIOMED
Based in Danvers, Massachusetts, USA, Abiomed, Inc. is a leading provider of medical devices that provide circulatory support. Our products are designed to enable the heart to rest by improving blood flow and/or performing the pumping of the heart. For additional information, please visit: www.abiomed.com. Abiomed, Impella, Impella 2.5, Impella 5.0, Impella 5.5, Impella LD, Impella CP, Impella RP, SmartAssist and Impella Connect are registered trademarks of Abiomed, Inc., and are registered in the U.S. and certain foreign countries. Impella BTR, Impella ECP, CVAD Study and STEMI DTU Study are pending trademarks of Abiomed, Inc.
FORWARD-LOOKING STATEMENTS
This release contains forward-looking statements, including statements regarding development of Abiomed's existing and new products, the company's progress toward commercial growth, and future opportunities and expected regulatory approvals. The company's actual results may differ materially from those anticipated in these forward-looking statements based upon a number of factors, including uncertainties associated with the scope, scale and duration of the impact of the COVID-19 pandemic, development, testing and related regulatory approvals, including the potential for future losses, complex manufacturing, high quality requirements, dependence on limited sources of supply, competition, technological change, government regulation, litigation matters, future capital needs and uncertainty of additional financing, and other risks and challenges detailed in the company's filings with the Securities and Exchange Commission, including the most recently filed Annual Report on Form 10-K and the filings subsequently filed with or furnished to the SEC. Readers are cautioned not to place undue reliance on any forward-looking statements, which speak only as of the date of this release. The company undertakes no obligation to publicly release the results of any revisions to these forward-looking statements that may be made to reflect events or circumstances that occur after the date of this release or to reflect the occurrence of unanticipated events.
View source version on businesswire.com: https://www.businesswire.com/news/home/20201016005123/en/
Contacts:
Director of Communication
(978) 882-8408
TLangford@abiomed.com
